Tutorial¶
Bio-specific features¶
Seq’s bio module contains all the following functions, types, and methods. The code snippets below should be preceded with from bio import *, although we omit that line for simplicity below.
Genomic types¶
Seq’s namesake type is indeed the sequence type: seq. A seq object represents a DNA sequence of any length and—on top of general-purpose string functionality—provides methods for performing common sequence operations such as splitting into subsequences, reverse complementation and \(k\)-mer extraction. Alongside the seq type are \(k\)-mer types, where e.g. Kmer[1] represents a 1-mer, Kmer[2] a 2-mer and so on, up to Kmer[1024].
Sequences can be seamlessly converted between these various types:
dna = s'ACGTACGTACGT' # sequence literal
# (a) split into subsequences of length 3
# with a step of 2
for sub in dna.split(k=3, step=2):
print(sub)
print(~sub) # reverse complement
# (b) split into 5-mers with step 1 (default)
for kmer in dna.kmers(k=5):
print(kmer)
print(~kmer) # reverse complement
# (c) convert entire sequence to 12-mer
kmer = Kmer[12](dna)
Seq also supports a pseq type for protein sequences:
protein = p'HEAGAWGHE' # pseq literal
print(list(protein.split(3, 3))) # [HEA, GAW, GHE]
print(s'ACCATGACA' |> translate) # TMT
Note
What’s the difference between sequences and \(k\)-mers in Seq? Sequences have arbitrary length and allow for ambiguous bases like N. \(k\)-mers, on the other hand, have a length that is fixed and must be known at compile time, only allowing for ACGT bases. \(k\)-mers can therefore be represented internally as 2-bit encoded integers, making them compact and very efficient to manipulate.
In practice, sequences would be read from e.g. a FASTQ file:
for record in FASTQ('input.fq'):
print('processing', record.name)
process(record.seq)
If you only care about the sequences, you can also do this:
for read in FASTQ('input.fq') |> seqs:
process(read)
Common formats like FASTQ, FASTA, SAM, BAM and CRAM are supported. The FASTQ and FASTA parsers support several additional options:
validate(Trueby default): Perform data validation as sequences are readgzip(Trueby default): Perform I/O using zlib, supporting gzip’d files (note that plain text files will still work with this enabled)fai(Trueby default; FASTA only): Look for a.faifile to determine sequence lengths before reading
For example:
for read in FASTQ('input.fq', validate=False, gzip=False) |> seqs:
process(read)
To read protein sequences, you can use pFASTA, which has the same interface as FASTA (but does not support fai):
for p in pFASTA('input.fa') |> seqs:
process(p)
Sequence matching¶
Seq provides the conventional match construct, which works on integers, lists, strings and tuples. Here’s a simple example:
def describe(n: int):
match n:
case m if m < 0:
print('negative')
case 0:
print('zero')
case m if 0 < m < 10:
print('small')
case _:
print('large')
A novel aspect of Seq’s match statement is that it also works on sequences, and allows for concise recursive representations of several sequence operations such as subsequence search, reverse complementation tests and base counting, as shown in this example:
# (a)
def has_spaced_acgt(s: seq):
match s:
case 'A_C_G_T*':
return True
case t if len(t) >= 8:
return has_spaced_acgt(s[1:])
case _:
return False
# (b)
def is_own_revcomp(s: seq):
match s:
case 'A*T' or 'T*A' or 'C*G' or 'G*C':
return is_own_revcomp(s[1:-1])
case s'':
return True
case _:
return False
# (c)
@tuple
class BaseCount:
A: int
C: int
G: int
T: int
def __add__(self, other: BaseCount):
a1, c1, g1, t1 = self
a2, c2, g2, t2 = other
return (a1 + a2, c1 + c2, g1 + g2, t1 + t2)
def count_bases(s):
match s:
case 'A*': return count_bases(s[1:]) + (1,0,0,0)
case 'C*': return count_bases(s[1:]) + (0,1,0,0)
case 'G*': return count_bases(s[1:]) + (0,0,1,0)
case 'T*': return count_bases(s[1:]) + (0,0,0,1)
case _: return BaseCount(0,0,0,0)
Example (a) checks if a given sequence contains the subsequence
A_C_G_T, where_is a wildcard base.Example (b) checks if the given sequence is its own reverse complement.
Example (c) counts how many times each base appears in the given sequence.
Sequence patterns consist of literal ACGT characters, single-base wildcards (_) or “zero or more” wildcards (...) that match zero or more of any base.
Pipelines¶
Pipelining is a natural model for thinking about processing genomic data, as sequences are typically processed in stages (e.g. read from input file, split into \(k\)-mers, query \(k\)-mers in index, perform full dynamic programming alignment, output results to file), and are almost always independent of one another as far as this processing is concerned. Because of this, Seq supports a pipe operator: |>, similar to F#’s pipe and R’s magrittr (%>%).
Pipeline stages in Seq can be regular functions or generators. In the case of standard functions, the function is simply applied to the input data and the result is carried to the remainder of the pipeline, akin to F#’s functional piping. If, on the other hand, a stage is a generator, the values yielded by the generator are passed lazily to the remainder of the pipeline, which in many ways mirrors how piping is implemented in Bash. Note that Seq ensures that generator pipelines do not collect any data unless explicitly requested, thus allowing the processing of terabytes of data in a streaming fashion with no memory and minimal CPU overhead.
Here’s an example of pipeline usage, which shows the same two loops from above, but as pipelines:
dna = s'ACGTACGTACGT' # sequence literal
# (a) split into subsequences of length 3
# with a stride of 2
dna |> split(..., k=3, step=2) |> print
# (b) split into 5-mers with stride 1
def f(kmer):
print(kmer)
print(~kmer)
dna |> kmers(k=5, step=1) |> f
First, note that split is a Seq standard library function that takes three arguments: the sequence to split, the subsequence length and the stride; split(..., k=3, step=2) is a partial call of split that produces a new single-argument function f(x) which produces split(x, k=3, step=2). The undefined argument(s) in a partial call can be implicit, as in the second example: kmers (also a standard library function) is parameterized by the target \(k\)-mer type and takes as arguments the sequence to \(k\)-merize, the \(k\)-mer length, and the stride; since just two of the three arguments are provided, the first is implicitly replaced by ... to produce a partial call (i.e. the expression is equivalent to kmers(..., k=5, step=1)). Both split and kmers are themselves generators that yield subsequences and \(k\)-mers respectively, which are passed sequentially to the last stage of the enclosing pipeline in the two examples.
Caution
The Seq compiler may perform optimizations that change the order of elements passed through a pipeline. Therefore, it is best to not rely on order when using pipelines. If order needs to be maintained, consider using a regular loop or passing an index alongside each element sent through the pipeline.
Sequence alignment¶
Aligning sequences is very straightforward in Seq, and supports numerous options/variants:
# default parameters
s1 = s'CGCGAGTCTT'
s2 = s'CGCAGAGTT'
aln = s1 @ s2
print(aln.cigar, aln.score)
# custom parameters
# match = 2; mismatch = 4; gap1(k) = 2k + 4; gap2(k) = k + 13
aln = s1.align(s2, a=2, b=4, gapo=4, gape=2, gapo2=13, gape2=1)
print(aln.cigar, aln.score)
Here is the list of options supported by the align() method; all are optional (default is global alignment):
a: match scoreb: mismatch scoreambig: ambiguous (i.e. N) match scoregapo: gap open costgape: gap extension costgapo2: 2nd gap open cost for dual gap cost functiongape2: 2nd gap extension cost for dual gap cost functionbandwidth: bandwidth for DP alignmentzdrop: off-diagonal drop-off to stop extensionscore_only: if true, don’t compute CIGARright: if true, right-align gapsapprox_max: if true, approximate maxapprox_drop: if true, approximate Z-droprev_cigar: if true, reverse CIGAR in outputext_only: if true, perform extension alignmentsplice: if true, perform spliced alignment
Note that all costs/scores are positive by convention.
Inter-sequence alignment¶
Seq uses ksw2 as its default alignment kernel. ksw2 does a good job of applying SIMD parallelization to align a single pair of sequences, which is referred to as intra-sequence alignment. However, we can often get better performance by aligning multiple sequences at once, referred to as inter-sequence alignment. Inter-sequence alignment is usually more cumbersome to program in general-purpose languages because many sequences need to be batched before performing the alignment. However, in Seq, inter-sequence alignment is as easy as intra-sequence, using the @inter_align annotation:
@inter_align
def process(t):
query, target = t
score = query.align(target, a=1, b=2, ambig=0, gapo=2, gape=1, zdrop=100, bandwidth=100, end_bonus=5)
print(query, target, score)
zip(seqs('queries.txt'), seqs('targets.txt')) |> process
Internally, the Seq compiler performs pipeline transformations when sequence alignment is performed within a function tagged @inter_align, so as to suspend execution of the calling function, batch sequences that need to be aligned, perform inter-sequence alignment and return the results to the suspended functions. Note that the inter-sequence alignment kernel used by Seq is adapted from BWA-MEM2.
Genomic index prefetching¶
Large genomic indices—ranging from several to tens or even hundreds of gigabytes—used in many applications result in extremely poor cache performance and, ultimately, a substantial fraction of stalled memory-bound cycles. For this reason, Seq performs pipeline optimizations to enable data prefetching and to hide memory latencies. You, the user, must provide just:
a
__prefetch__magic method definition in the index class, which is logically similar to__getitem__(indexing construct) but performs a prefetch instead of actually loading the requested value (and can simply delegate to__prefetch__methods of built-in types);a one-line
@prefetchannotation on functions that should perform prefetching.
In particular, a typical prefetch-friendly index class would look like this:
class MyIndex: # abstract k-mer index
...
def __getitem__(self, kmer: Kmer[20]):
# standard __getitem__
def __prefetch__(self, kmer: Kmer[20]):
# similar to __getitem__, but performs prefetch
Now, if we were to process data in a pipeline as such:
@prefetch
def process(read: seq, index: MyIndex):
...
for kmer in read.kmers(k=20, step=step):
hits_fwd = index[kmer]
hits_rev = index[~kmer]
...
return x
FASTQ("reads.fq") |> seqs |> process(index) |> postprocess
The Seq compiler will perform pipeline transformations to overlap cache misses in MyIndex with other useful work, increasing overall throughput. In our benchmarks, we often find these transformations to improve performance by 50% to 2×. However, the improvement is dataset- and application-dependent (and can potentially even decrease performance, although we rarely observed this), so users are encouraged to experiment with it for their own use case.
As a concrete example, consider Seq’s built-in FM-index type, FMIndex, and a toy application that counts occurences of 20-mers from an input FASTQ. FMIndex provides end-to-end search methods like locate() and count(), but we can take advantage of Seq’s prefetch optimization by working with FM-index intervals:
from bio.fmindex import FMIndex
fmi = FMIndex('/path/to/genome.fa')
k, step, n = 20, 20, 0
def update(count: int):
global n
n += count
@prefetch
def find(s: seq, fmi: FMIndex):
intv = fmi.interval(s[-1]) # initial FM-index interval
s = s[:-1] # trim off last base of sequence
while s and intv:
intv = fmi[intv, s[-1]] # backwards extend FM-index interval
s = s[:-1] # trim off last base of sequence
return len(intv) # return count of sequence in index
FASTQ('/path/to/reads.fq') |> seqs |> split(k, step) |> find(fmi) |> update
print('total:', n)
That single @prefetch line can have a significant impact, especially for larger k. Here is a graph of the performance of this exact snippet for various k using hg19 as the reference:
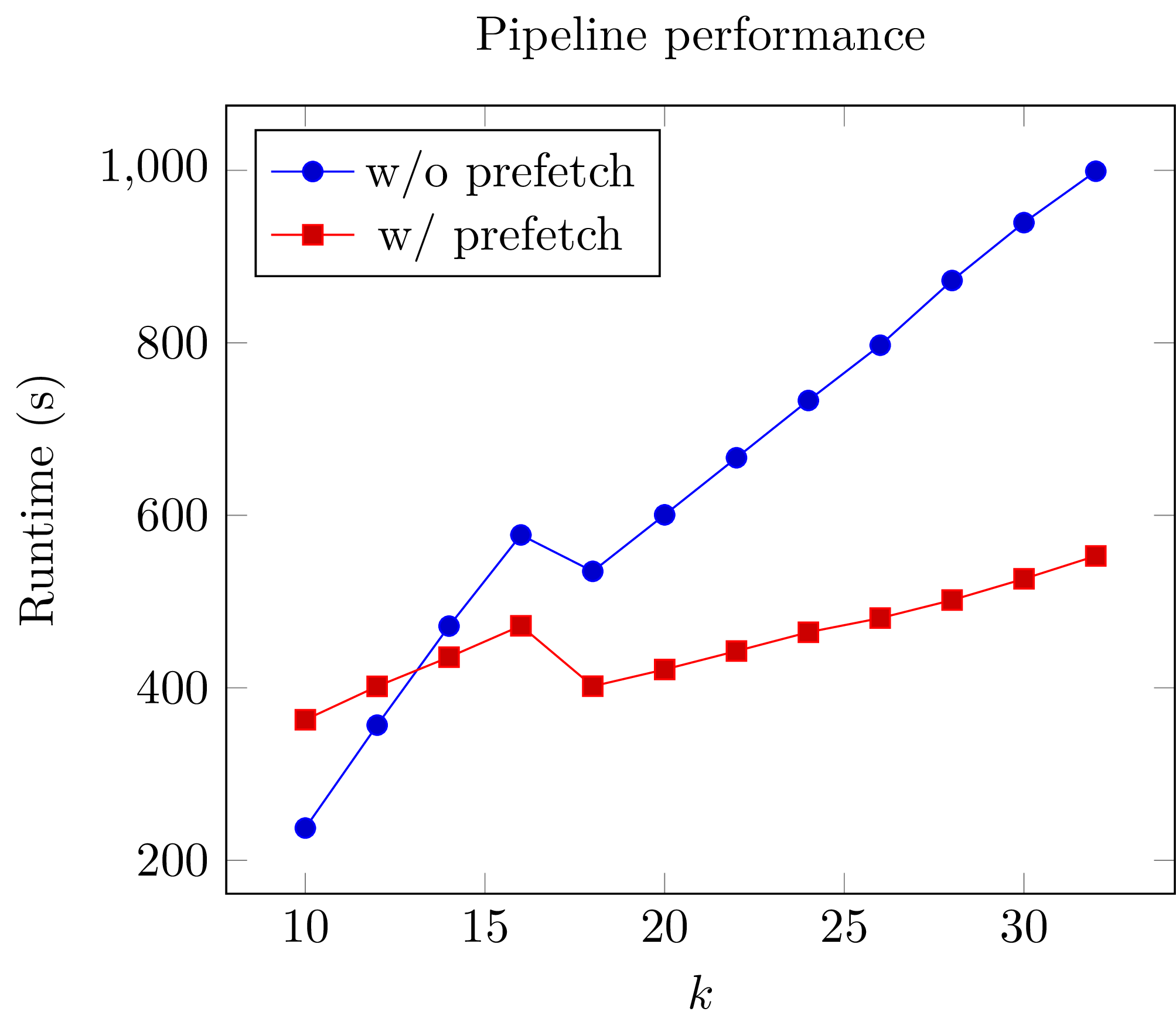
Other features¶
Parallelism¶
CPython and many other implementations alike cannot take advantage of parallelism due to the infamous global interpreter lock, a mutex that protects accesses to Python objects, preventing multiple threads from executing Python bytecode at once. Unlike CPython, Seq has no such restriction and supports full multithreading. To this end, Seq supports a parallel pipe operator ||>, which is semantically similar to the standard pipe operator except that it allows the elements sent through it to be processed in parallel by the remainder of the pipeline. Hence, turning a serial program into a parallel one often requires the addition of just a single character in Seq. Further, a single pipeline can contain multiple parallel pipes, resulting in nested parallelism. As an example, here are the same two pipelines as above, but parallelized:
dna = s'ACGTACGTACGT' # sequence literal
# (a) split into subsequences of length 3
# with a stride of 2
dna |> split(..., k=3, step=2) ||> print
# (b) split into 5-mers with stride 1
def f(kmer):
print(kmer)
print(~kmer)
dna |> kmers(k=5, step=1) ||> f
Internally, the Seq compiler uses an OpenMP task backend to generate code for parallel pipelines. Logically, parallel pipe operators are similar to parallel-for loops: the portion of the pipeline after the parallel pipe is outlined into a new function that is called by the OpenMP runtime task spawning routines (as in #pragma omp task in C++), and a synchronization point (#pragma omp taskwait) is added after the outlined segment. Lastly, the entire program is implicitly placed in an OpenMP parallel region (#pragma omp parallel) that is guarded by a “single” directive (#pragma omp single) so that the serial portions are still executed by one thread (this is required by OpenMP as tasks must be bound to an enclosing parallel region).
Type extensions¶
Seq provides an @extend annotation that allows programmers to add and modify methods of various types at compile time, including built-in types like int or str. This actually allows much of the functionality of built-in types to be implemented in Seq as type extensions in the standard library. Here is an example where the int type is extended to include a to method that generates integers in a specified range, as well as to override the __mul__ magic method to “intercept” integer multiplications:
@extend
class int:
def to(self, other: int):
for i in range(self, other + 1):
yield i
def __truediv__(self, other: int):
print('caught int div!')
return 42
for i in (5).to(10):
print(i) # 5, 6, ..., 10
# prints 'caught int div!' then '42'
print(2 / 3)
Note that all type extensions are performed strictly at compile time and incur no runtime overhead.
Other types¶
Seq provides arbitrary-width signed and unsigned integers up to Int[512] and UInt[512], respectively (note that int is an Int[64]). Typedefs for common bit widths are provided in the standard library, such as i8, i16, u32, u64 etc.
The Ptr[T] type in Seq also corresponds to a raw C pointer (e.g. Ptr[byte] is equivalent to char* in C). The array[T] type represents a fixed-length array (essentially a pointer with a length).
Seq also provides __ptr__ for obtaining a pointer to a variable (as in __ptr__(myvar)) and __array__ for declaring stack-allocated arrays (as in __array__[int](10)).
Calling BWA from Seq¶
Seq provides a built-in module for interfacing with BWA. To use this module, simply build a shared BWA library and set BWA_LIB accordingly:
git clone https://github.com/lh3/bwa
cd bwa
make
gcc -shared -o libbwa.so *.o -lz
export BWA_LIB=`pwd`/libbwa.so
Now BWA can be used in Seq as such:
# Implementation of https://github.com/lh3/bwa/blob/master/example.c
from sys import argv
from bio.bwa import *
bwa = BWA(argv[1])
for read in FASTQ(argv[2]):
for reg in bwa.align(read.read):
if reg.secondary >= 0: continue
aln = bwa.reg2aln(read.read, reg)
print(read.name, '-' if aln.rev else '+', bwa.name(aln), aln.pos, aln.mapq, aln.cigar, aln.NM)
This program can be invoked as seqc run example.seq /path/to/hg19.fa /path/to/reads.fq.
BWA options can be passed via BWA(options(...), ...). For example, to set a mismatch score of 5, use BWA(options(mismatch_score=5), "hg19.fa"). Valid options are:
match_scoremismatch_scoreopen_delopen_insextend_delextend_insbandwidthzdropclip_penaltyunpaired_penalty
Consult the BWA documentation for a detailed description of each of these.